How can I produce a multiple sequence alignment of TB sequences?
Learning Objectives:
Use nf-core/bactmap to produce a multiple sequence alignment.
Keypoints:
To build a phylogenetic tree we need a multiple sequence alignment of the sequences we want to infer a tree from.
Alignments are usually done against a reference genome.
We can use the pipeline nf-core/bactmap to produce a multiple sequence alignment.
9.1 nf-core/bactmap
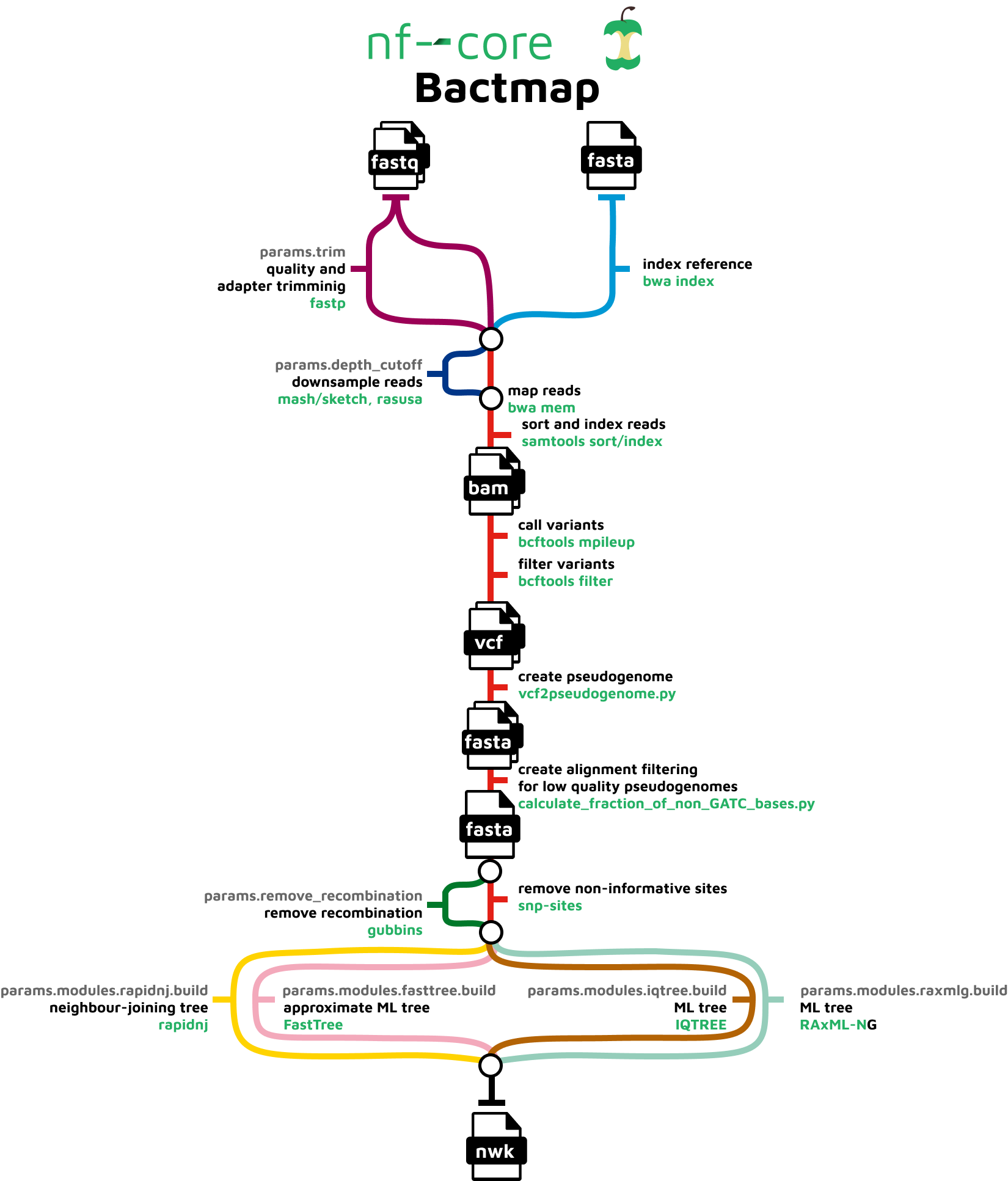
nf-core/bactmap is a bioinformatics best-practice analysis pipeline for mapping short reads from bacterial WGS to a reference sequence, creating filtered VCF files, making pseudogenomes based on high quality positions in the VCF files and optionally creating a phylogeny from an alignment of the pseudogenomes. It makes use of many of the tools and techniques we’ve covered so far this week.
nf-core/bactmap variant calling pipeline diagram from nf-core (https://nf-co.re/bactmap).
9.2 TB dataset
We will be analysing a dataset of 83 TB genomes previously analysed by Prince as part of his PhD. As we don’t have access to the Noguchi HPC yet, we’re only going to analyse two of the genomes for now. Don’t worry, we’ll go through the results for all 83 samples
9.3 Activate the nextflow environment
Now navigate into the 09-bactmap/ directory and activate the nextflow environment:
cd ~/Desktop/workshop_files_Bact_Genomics_2023/09-bactmap/
mamba activate nextflow
Disk Usage I — Before analysis
Before we run nf-core/bactmap, let’s pause and check the space of our current working directory as we did for our previous lesson.
You can do this with the disk usage du command
du-h
Current Disk Space In 09-bactmap Directory
~247MB
Now, keep this value in mind, and this time, don’t forget it. We will come back to it at the end of this chapter.
9.4 The MTBC ancestral sequence
The reference sequence we’ll use to map our TB samples back to is one commonly used in the MTBC community and is known as the MTBC ancestral sequence. It was generated as part of Comas et al. 2013 (link to paper). This sequence was created by building an alignment and phylogenetic tree of the known MTBC genomes at the time and inferring what the most likely sequence of the ancestor of the MTBC was. It is a useful sequence to use when comparing different members of the MTBC as it should reduce the amount of bias towards any particular reference that is more closely related to one lineage than another.
9.5 Setting up the Nextflow config file
As we learnt in the nf-core pipelines module, we can create a config file that tells Nextflow which parameters to use e.g. the job submission system. We’ve create a basic template that will allow you to run nf-core/bactmap on the teaching computers/HPC. The config file looks like this:
The first step in building a phylogenetic tree is to generate a multiple sequence alignment. To do this, we’re going to map the sequence data for our 83 TB genomes to a reference, in this case the MTBC inferred ancestral sequence, using the nf-core/bactmap pipeline.
Generate a samplesheet
Now create a samplesheet.csv file containing the sample IDs and the location of the files to be mapped:
nf-core/bactmap has a number of optional arguments but for now these are the ones we’re going to use:
Input option
Description
-profile conda
Create conda environments for each tool in the pipeline (we could also use Singularity or Docker)
-c teaching.conf
A confuguration file containing the settings needed to run the pipeline on your machines
--input samplesheet_test.csv
The samplesheet we created above containing the sample ids and location of the fastq files
--reference reference/MTB_ANC_unwrapped.fasta
The reference sequence we’re going to map our samples to
--genome_size 4.3mb
This is used by the pipeline to calculate the approximate genome coverage in the fastq files. By default the pipeline uses a tool called rasusa to subsample the fastq files so the genome coverage is <= 100X
--outdir results/bactmap
The directory we’re going to save the outputs from nf-core/bactmap to
Visit the nf-core/bactmap page for further information on running the pipeline with different options.
If the pipeline has started successfully, you should see something like this:
N E X T F L O W ~ version 22.04.3Launching `https://github.com/nf-core/bactmap` [special_kare] DSL2 - revision: e83f8c5f0e [master]------------------------------------------------------,--./,-. ___ __ __ __ ___ /,-._.--~'|\ ||__ __ / ` / \ |__)|__ }{| \|| \__, \__/| \ |___ \`-._,-`-, `._,._,' nf-core/bactmap v1.0.0------------------------------------------------------Core Nextflow options revision : master runName : special_kare containerEngine : singularity launchDir :/rds/project/rds-PzYD5LltalA/Teaching/Ghana workDir :/rds/project/rds-PzYD5LltalA/Teaching/Ghana/work projectDir :/home/ajv37/.nextflow/assets/nf-core/bactmap userName : ajv37 profile : singularity configFiles :/home/ajv37/.nextflow/assets/nf-core/bactmap/nextflow.config,/rds/project/rds-PzYD5LltalA/Teaching/Ghana/cambridge.configInput/output options input : samplesheet.csv outdir : bactmap_resultsCompulsory parameters reference : MTB_ANC_unwrapped.fastaOptional pipeline steps adapter_file :/home/ajv37/.nextflow/assets/nf-core/bactmap/assets/adapters.fas genome_size :4.3mbMax job request options max_cpus :56 max_memory :192 GB max_time :12hInstitutional config options config_profile_description: Cambridge HPC cluster profile. config_profile_contact : Andries van Tonder(ajv37@cam.ac.uk) config_profile_url : https://docs.hpc.cam.ac.uk/hpc!! Only displaying parameters that differ from the pipeline defaults !!------------------------------------------------------If you use nf-core/bactmap for your analysis please cite:* The nf-core framework https://doi.org/10.1038/s41587-020-0439-x* Software dependencies https://github.com/nf-core/bactmap/blob/master/CITATIONS.md------------------------------------------------------executor >slurm(2)executor >slurm(2)[3f/931dc1] process > NFCORE_BACTMAP:BACTMAP:INPUT_CHECK:SAMPLESHEET_CHECK(samplesheet.csv)[0%]0 of 1[59/74ef4b] process > NFCORE_BACTMAP:BACTMAP:BWA_INDEX(MTB_ANC_unwrapped.fasta)[0%]0 of 1[-] process > NFCORE_BACTMAP:BACTMAP:FASTP -[-] process > NFCORE_BACTMAP:BACTMAP:SUB_SAMPLING:RASUSA -[-] process > NFCORE_BACTMAP:BACTMAP:BWA_MEM -[-] process > NFCORE_BACTMAP:BACTMAP:BAM_SORT_SAMTOOLS:SAMTOOLS_SORT -[-] process > NFCORE_BACTMAP:BACTMAP:BAM_SORT_SAMTOOLS:SAMTOOLS_INDEX -[-] process > NFCORE_BACTMAP:BACTMAP:BAM_SORT_SAMTOOLS:BAM_STATS_SAMTOOLS:SAMTOOLS_...-[-] process > NFCORE_BACTMAP:BACTMAP:BAM_SORT_SAMTOOLS:BAM_STATS_SAMTOOLS:SAMTOOLS_...-[-] process > NFCORE_BACTMAP:BACTMAP:BAM_SORT_SAMTOOLS:BAM_STATS_SAMTOOLS:SAMTOOLS_...-[-] process > NFCORE_BACTMAP:BACTMAP:VARIANTS_BCFTOOLS:BCFTOOLS_MPILEUP -[-] process > NFCORE_BACTMAP:BACTMAP:VARIANTS_BCFTOOLS:BCFTOOLS_FILTER -[-] process > NFCORE_BACTMAP:BACTMAP:VCF2PSEUDOGENOME -[-] process > NFCORE_BACTMAP:BACTMAP:ALIGNPSEUDOGENOMES -[-] process > NFCORE_BACTMAP:BACTMAP:SNPSITES -[-] process > NFCORE_BACTMAP:BACTMAP:GET_SOFTWARE_VERSIONS -[-] process > NFCORE_BACTMAP:BACTMAP:MULTIQC -
The pipeline will take a while to run so we’ll have a look at the results after lunch.