6.1 Assembly and Annotation
Teaching: 50 min || Exercises: 20 min
Overview
6.1 Background
There are two approaches for genome assembly: reference-based (or comparative) or de novo. In a reference-based assembly, we use a reference genome as a guide to map our sequence data to and thus reassemble our sequence this way (this is what we did in the previous module). Alternatively, we can create a ‘new’ (de novo) assembly that does not rely on a map or reference and more closely reflects the actual genome structure of the isolate that was sequenced.
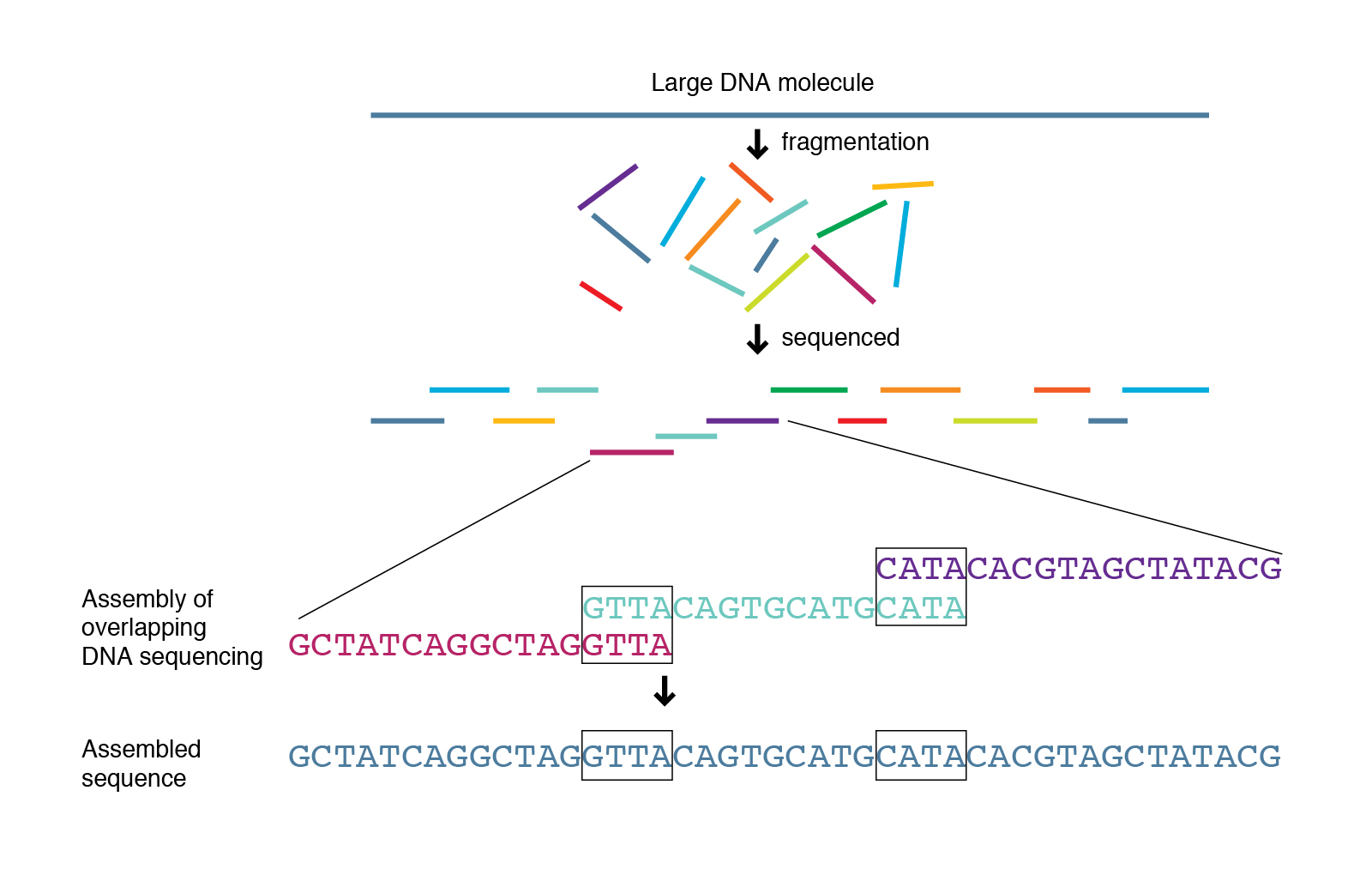
Genome assemblers
Several tools are available for de novo genome assembly depending on whether you’re trying to assemble short-read sequence data, long reads or else a combination of both. Two of the most commonly used assemblers for short-read Illumina data are Velvet and SPAdes. SPAdes has become the de facto standard de novo genome assembler for Illumina whole genome sequencing data of bacteria and is a major improvement over previous assemblers like Velvet. However, some of its components can be slow and it traditionally did not handle overlapping paired-end reads well. Shovill is a pipeline which uses SPAdes at its core, but alters the steps before and after the primary assembly step to get similar results in less time. Shovill also supports other assemblers like SKESA, Velvet and Megahit.
Disk Usage I — Before analysis
Before we start performing any assemblies, let’s pause and check the space of our current working directory as we did for our previous lesson.
You can do this with the disk usage du command
du -hCurrent Disk Space In assembly_annotation_MTB Directory
~247MB6.2 de novo genome assembly
de novo genome assembly with Shovill
We’re going to use Shovill to create our de novo genome assemblies. Let’s navigate the module directory and activate the shovill environment:
cd workshop_files_Bact_Genomics_2023/06_assembly_annotation
mamba activate shovillWe’ve chosen one of the TB genomes from our course dataset to assemble with Shovill: ERX1275297_ERR1203055 (TBNm 2359).
Before we move on to the next step, let’s rename our assembly (contigs.fa) to something more useful:
cd ERX1275297_ERR1203055
mv contigs.fa ERX1275297_ERR1203055_contigs.faFinally, deactivate the shovill environment:
mamba deactivate6.3 Assembly quality assessment
Assembly QC with QUAST
Before we do any further analyses with our assemblies, we need to assess the quality. To do this we use a tool called QUAST which generates a number of useful metrics for assemblies. These include but aren’t limited to:
- The total number of contigs greater than 0 bp. Ideally, we like to see assemblies in the smallest number of contigs (‘pieces’) as this means there is likely less missing data contained in gaps between contigs.
- The total length of the assembly. We normally know what the expected length of our assembly is (for more diverse organisms like E. coli there may be a range of genome sizes). The length of your assembly should be close to the expected genome size. If it’s too big or too small, either there is some kind of contamination or else you’ve sequenced the wrong species!
- The N50 of the assembly. This is the final metric often used to assess the quality of an assembly. The N50 is typically calculated by averaging the length of the largest contigs that account for 50% of the genome size. This is a bit more complicated conceptually but the higher the N50 the better. A large N50 implies that you have a small number of larger contigs in your assembly which equals a good assembly.
Let’s activate the quast environment:
mamba activate quastFinally, deactivate the quast environment:
mamba deactivate6.4 Genome annotation
Genome annotation is a multi-level process that includes prediction of protein-coding genes (CDSs), as well as other functional genome units such as structural RNAs, tRNAs, small RNAs, pseudogenes, control regions, direct and inverted repeats, insertion sequences, transposons and other mobile elements. The most commonly used tools for annotating bacterial genomes are Prokka and, more recently, Bakta. Both use a tool called prodigal to predict the protein-coding regions along with other tools for predicting other genomic features such as Aragorn for tRNA. Once the genomic regions have been predicted, the tools use a database of existing bacterial genome annotations, normally generated from a large collection of genomes such as UniRef, to add this information to your genome.
Annotation with Prokka
Prokka is a software tool to annotate bacterial, archaeal and viral genomes quickly and produce standards-compliant output files (GenBank, EMBL and gff) for further analysis or viewing in genome browsers. It is suitable for annotating de novo assemblies of bacteria, but not appropriate for human genomes (or any other eukaryote).
Before we run Prokka, let’s activate the environment:
mamba activate prokkaThe file format most commonly used for downstream analyses such as manual curation or pan-genome analysis is the gff file. We covered gff files back in Common File Formats so you may want to remind yourself what’s inside them.
Finally, deactivate the prokka environment:
mamba deactivateDisk Usage II — Cleaning up after analysis
Now that we are done investigating our assembling and annotating our genome, let’s pause again and check the space of our current working directory.
You can do this with the disk usage du command
du -hHow much disk space have you used since the start of the analysis?
Credit
Information on this page has been adapted and modified from the following source(s): https://github.com/Joseph7e/MDIBL-T3-WGS-Tutorial#genome-assembly